The ability to simulate the behavior of systems at the atomic level represents a powerful tool for everything from drug design to materials discovery. A team led by Los Alamos National Laboratory researchers has developed machine learning interatomic potentials that predict molecular energies and forces acting on atoms, enabling simulations that save time and expense compared with existing computational methods.
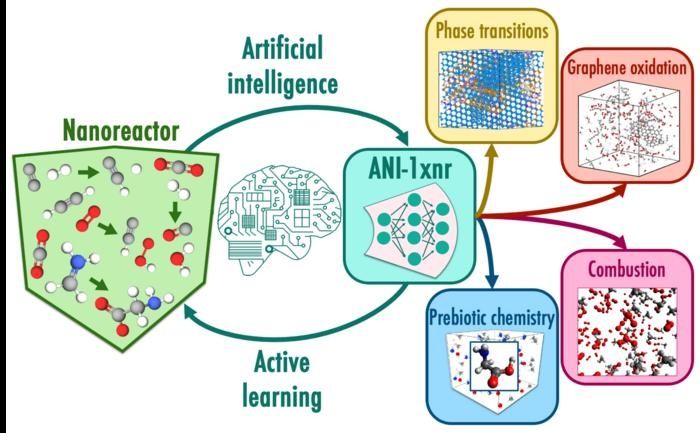
In this workflow, nanoreactor simulations automatically sample reactive chemical space without relying on human intuition. The nanoreactor is a special class of atomistic simulations in which chemical reactions are induced by colliding molecules at high velocities. Active learning utilizes the machine learning potential, ANI-1xnr, to drive the nanoreactor dynamics and sub-select structures with high uncertainties. Case studies such as carbon phase transitions of carbon and methane combustion test the generality of the resulting model, ANI-1xnr.
Los Alamos National Laboratory
“Machine learning potentials increasingly offer an effective alternative to computationally expensive simulations that try to represent complex physical systems on the atomic scale,” said Benjamin Nebgen, Los Alamos chemical physicist and co-author of a recent Nature Chemistry paper describing the work. “A general reactive machine learning interatomic potential, applicable to a broad range of reactive chemistry without the need for refitting, will greatly benefit chemistry and materials science.”
Bridging the gap in effective simulations
Building effective simulations for molecular dynamics in chemistry is traditionally done with physics-based computational models, including classical force fields or quantum mechanics. While quantum mechanical models are accurate and generally applicable, they are extremely computationally expensive. By contrast, classical force fields are computationally efficient, but of relatively low accuracy and only applicable to a limited range of systems. ANI-1xnr, the team’s transformational machine learning model, bridges the gap in speed, accuracy and generality that has existed in chemistry for many decades. (Machine learning is an application of artificial intelligence where computer programs “learn” through training.)
ANI-1xnr represents the first reactive machine learning interatomic potential general enough — it can be applied to many different chemical systems — to compete with physics-based computational models for performing large-scale reactive atomistic simulations. ANI-1xnr was developed using an automated workflow that performed reactive molecular dynamics simulations over a wide range of chemical systems containing carbon, hydrogen, nitrogen and oxygen elements.
ANI-1xnr proved capable of studying a diverse range of systems, from carbon phase transitions to combustion to prebiotic chemistry. The team validated the simulations by comparing them with results from experiments and from conventional computational techniques.
A transformational interatomic potential
“ANI-1xnr does not require domain expertise or refitting for every new use case, enabling scientists from a diverse range of domains to study unknown chemistry,” said Richard Messerly, computational scientist at Los Alamos and co-corresponding author of the paper. “The general applicability of ANI-1xnr is transformational, representing a significant step toward replacing the long-standing modeling techniques for studying reactive chemistry at scale.”
The data set used by the team and the ANI-1xnr code has been made publicly available to the research community.
Original publication
Shuhao Zhang, Małgorzata Z. Makoś, Ryan B. Jadrich, Elfi Kraka, Kipton Barros, Benjamin T. Nebgen, Sergei Tretiak, Olexandr Isayev, Nicholas Lubbers, Richard A. Messerly, Justin S. Smith; “Exploring the frontiers of condensed-phase chemistry with a general reactive machine learning potential”; Nature Chemistry, 2024-3-7









{kind=link}